2022年2月10日,美国 FDA线上举行了针对信达生物、礼来制药联合研发的 PD-1抗体信迪利单抗上市申请的肿瘤药物咨询委员会(ODAC)。
出人意料的是,这次专家委员会的最终投票结果为14:1,认为信迪利单抗需要补充额外的临床试验,来证明自己在美国的适用性。

来源:ODAC审评文件
最引人注意的,还是 FDA引用了2016年中国食药监局披露的数据:80%的中国临床试验不标准。

来源:ODAC审评文件
信迪利单抗此次上市申请,是国产 PD-1药物首次出海。但在前期纵享丝滑之后,后期却迎来 FDA当头一棒——为什么出海卡在了审核关?又为什么投票如此悬殊?值得我们一看。
国产 PD-1的扬帆
2018年12月,由信达生物、礼来制药联合研发的信迪利单抗注射液(达伯舒)在中国获批上市,用于治疗复发或难治性经典型霍奇金淋巴瘤(R/R cHL)。
2019年11月,信迪利单抗以降价63%的代价,成为第一个进入国家医保目录的 PD-1产品。
受益于进入医保,信迪利单抗的覆盖范围由2019年底的约2000家医院和300间药房扩增到截至2020年12月31日的约4000家医院和900间药房。得益于此,2020年信迪利单抗的全年销售额达到22.90亿元,较前一年度增加125.4%。
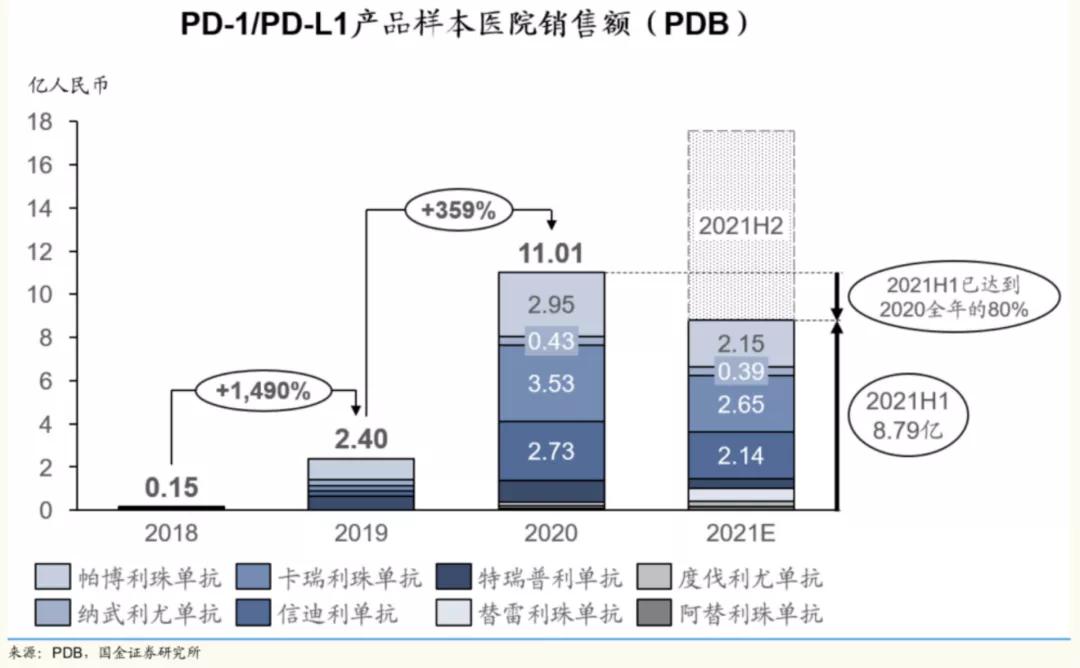
来源:PDB,国金证券研究所
次年3月,信达生物向 FDA递交了信迪利单抗的上市申请。
2021年5月17日,美国食品药品监督管理局(FDA)正式受理了信迪利单抗联合培美曲塞和铂类用于非鳞状非小细胞肺癌(NSCLC)一线治疗的上市申请。这也是目前中国首个自主研发,上市申请被 FDA受理并进入正式审评阶段的生物创新药。
然而,热板凳还没坐上多久,当受理上市申请快进到审核阶段,结果却是压倒性的“14:1”不通过。
折戟
先来看官方角度,在信达和礼来的这次折戟上,FDA给出了三个解释。
第一个是临床格局的变化。
FDA认为,2018年8月20日 Keytruda(帕博利珠单抗)联合化疗即获批用于治疗非鳞状 NSCLC,并且 K药临床数据已经在审批之前广泛宣传,但是信迪利单抗的 ORIENT-11实验在此之后才启动。
启动时,一线转移性肺癌的护理标准已经发生变化,临床一线疗法从化疗变为“免疫治疗+化疗”联合治疗。鉴于 Keytruda(帕博利珠单抗)化疗在临床和统计学上显示出对总生存期的显着益处,研究人员不应该将患者纳入化疗控制组。

来源:ODAC审评文件
第二件事,是 FDA认为单国临床研究数据不适用于美国患者。
FDA表示,ORIENT-11的研究人群完全由来自一个国家的亚洲患者组成。虽然中国是一个多民族和多民族的国家,但 ORIENT-11研究人群并未反映美国肺癌患者的种族和民族多样性。
“接受此类研究和类似研究,与行业范围内对临床试验公平代表性的承诺相冲突。”


来源:ODAC审评文件
第三个原因,是它选择了无进展生存时间(PFS)作为临床试验终点,而没有选择总生存期终点(OS)。
迄今为止,FDA对转移性非小细胞肺癌一线免疫治疗方案的所有批准都是基于 OS的统计学改善。总生存期被认为是最可靠的癌症终点,并且在可以合理评估时是首选。
所以,FDA表达了对 PFS后继续用药的担忧,表示 ORIENT-11在设计上显示缺乏多样性。
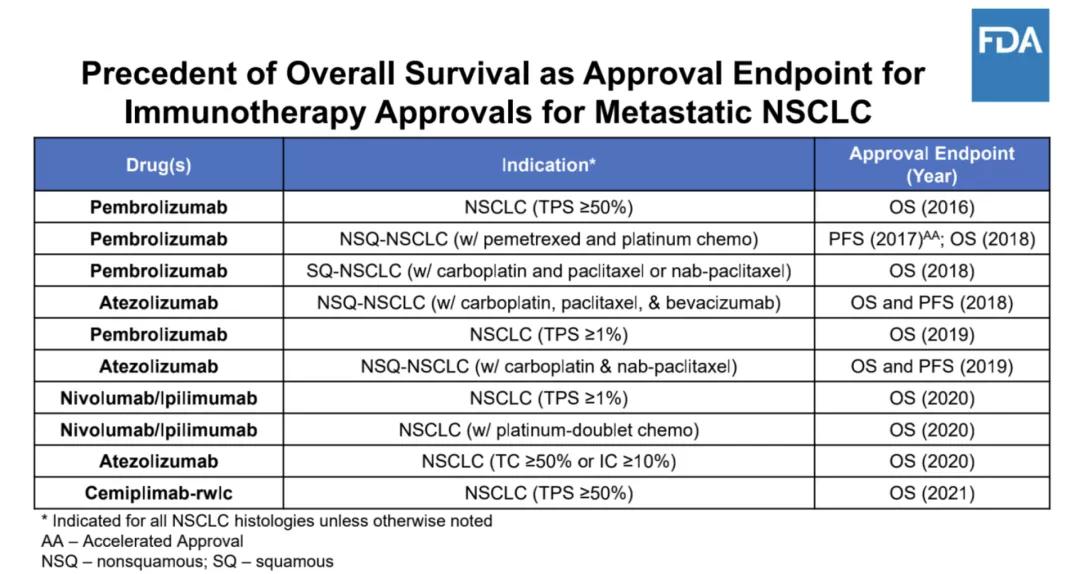
Image来源:ODAC审评文件
另外,FDA还指出,申请人一直没有就“研究设计或试验”进行咨询 FDA。如果咨询了 FDA,可能会建议将信迪利单抗与 FDA批准的具有总生存期终点(OS)的 PD-1药物进行头对头研究。
风向突变的 FDA
来拉一下时间线。
其实早在2019年,FDA肿瘤学卓越中心主任 Richard Pazdur就曾公开呼吁中国药企将国产 PD-1/PD-L1抑制剂引入美国市场。
“这对每个人来说都可能是一件好事,因为我们还没有看到西方制药公司在价格上有所调整。”他如是说道。
他当时还表示,中国公司模仿 FDA批准产品开发新的 PD-1/PD-L1药物,获得 FDA的批准将是没有太大悬念的,“显然,它们可能会给出非常相似的结果,所以我们在批准这些药物时,会很顺利。”
在此之后,信达、君实、康方、百济神州先后向 FDA递交了上市申请。
然而在2021年,Pazdur却突然转变态度。
2021年12月15日,Pazdur在 NEJM上发表文章 The Wild West of Checkpoint Inhibitor Development,指出已上市的 PD-1/PD-L1药物的适应症中的45%是通过加速审批(Accelerated Approval)这一途径获批的,通常是只做了单臂试验,之后随机临床试验可能显示出不一致的结果。
“用低于美国临床对照标准的方式,在中国开发的药,是不值得 FDA给予照顾来美国上市的。”
在会前一周的2022年2月4日,Pazdur又在柳叶刀上发表题为 Importing oncology trials from China: a bridge over troubled water的文章。
文章指出,至少有25个来自中国的免疫抑制剂类新药申请,都几乎只基于在中国做的临床试验数据,重申“单一国外数据不能代表美国人口”这一观点,与2019年那次医学会议上的公开评论大相径庭。
不管是疫情初期对中国外科口罩的变脸,还是之前对国产药物的“无差别抵制”,FDA出尔反尔已经不是第一次。这次的“渣男行为”,对想出海的药企而言,无疑是当头一棒,所以就连礼来也吃了一个暗亏。
Fast-Follow创新药
不过,从药物本身创新逻辑的角度来看,FDA这一次虽然不厚道,可话糙、理没那么糙。因为大部分国内所谓的创新药,不过是 Fast-Follow的产物。
Fast-Follow名为快速追踪新药模式,指在不侵犯他人专利的情况下,在已有靶点和机理的基础上,对新药进行分子结构改造或修饰,寻找作用机制相同或相似,具有新治疗效果的新药物。
Fast-Follow包括了 Me-too、Me-better、Me-worse等药物。
Me-too药顾名思义,即药物结构与首创药相似,只有较小差别,这个较小的差别区分了 Me-too药和仿制药。
与仿制药不同,仿制药完全照抄原研药,需要等原研药专利过期后才能投入市场。但是 Me-too药由于绕开了专利,即便原研药仍然在专利期也不需要授权。
Me-better指改良模仿,也就是在原研药基础上创新,得到比原研药更好的疗效。
Me-worse是指虽然绕开了原研药专利,但疗效并不如原研药,现在已经很难获得 NMPA批准。
如果创新是头发,那仿制药就是锃光瓦亮的秃头,Fast Follow药是三毛——秃了,但没完全秃。优势是既能用较少的时间金钱成本开发出新药,又能规避首创药的专利保护。
目前,所有的国产 PD-1抑制剂都属于 Me-too药。虽然所有的国内厂商都号称自己的药是 Me-better药,即药物经过改造后,临床效果要好于首创药,但可惜临床试验的结果并不能自证。
这也正是国内创新药市场的一个缩影——创新性不够。
已有的大部分创新药,都是基于热门靶点的研究,跟随首创药的“小打小闹”。在中国,同类药物想要打败原研药,可以依靠 Fast-Follow的成本优势,这其中,想要快速抢占市场更快的上市至为关键。
如果没有2019年的医保谈判,在“First-in-class”药厂吃肉之余,追随前者脚步的 Me-too药厂依靠喝汤也能活得十分惬意。但一轮又一轮的医保谈判就像一面“照妖镜”,药厂的创新能力在“照妖镜”面前一目了然。
于是药企又想出了一招——出海。
这次谁也没想到的是,反复无常的 FDA又给药企们上了一课。
市场竞争背后,患者才是主体
截至去年12月,NMPA已经批准了12款 PD-1/PD-L1产品,涉及11个癌种,44个适应症。其中非小细胞肺癌:MSD、恒瑞、百济神州、信达均获批了一线鳞癌和腺癌。
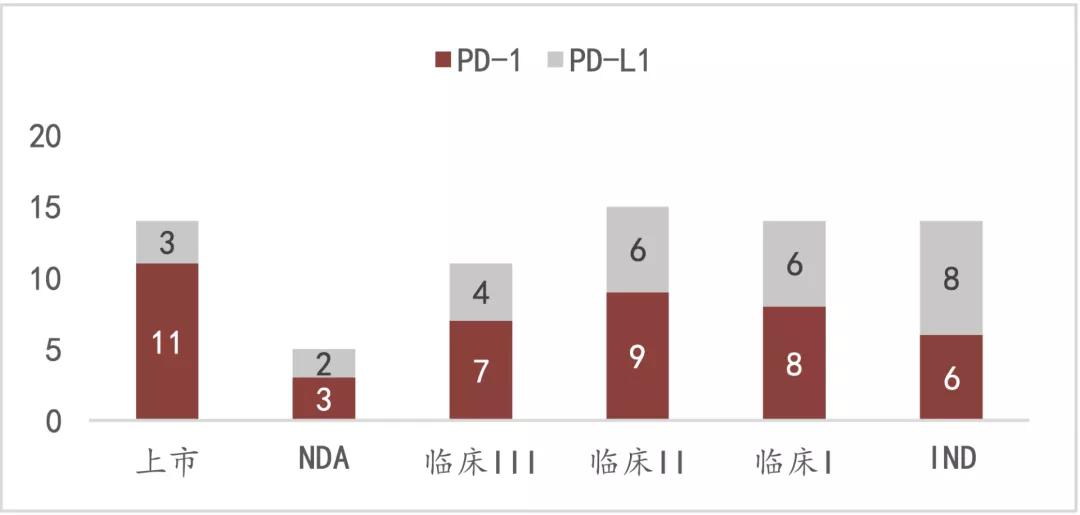
国内 PD-1/PD-L1临床研究数量来源:FIC intelligence, Insight,莫尼塔研究
PD-1的市场竞争,已经完全进入白热化阶段。
而国内 PD-(L)1抑制剂市场能否规避“内卷”的危害,已成为一个迫切需要回答的问题。
在目前已批准的44个适应证中,有25个来自国产 PD-(L)1抑制剂,其中16个是重复适应证,仅有9个为独家或同类首个获批适应证。
与之形成鲜明反差的是,在4个进口 PD-(L)1抑制剂获批的19个适应证中,有18个仍是独家或是同类首个获批适应证。
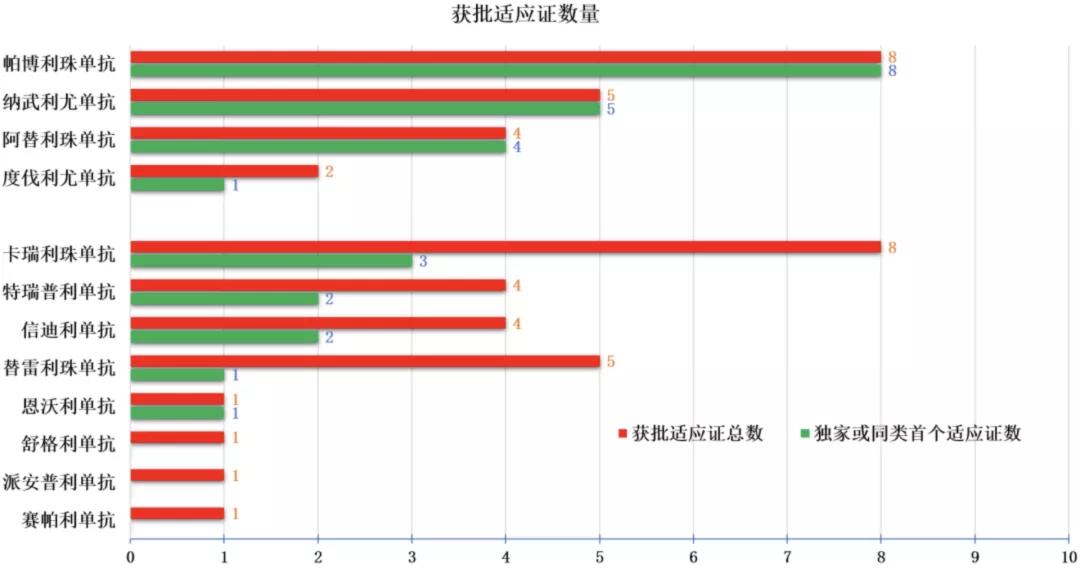
PD-(L)1抑制剂国内获批适应证数量及独家或首个适应证数量,“统计标准以获批受理号为准”,图片来源:文献整理
那些本就创新不足的国产 PD-(L)1抑制剂,所开展的新临床研究,能否满足患者之前未被满足的需求,将会是接下来药企们最需要交的答卷。







{kind=link}